
- Home
- News
- Analysis
- States
- Perspective
- Videos
- Education
- Entertainment
- Elections
- World Cup 2023
- Features
- Health
- Budget 2024-25
- Business
- Series
- NEET TANGLE
- Economy Series
- Earth Day
- Kashmir’s Frozen Turbulence
- India@75
- The legend of Ramjanmabhoomi
- Liberalisation@30
- How to tame a dragon
- Celebrating biodiversity
- Farm Matters
- 50 days of solitude
- Bringing Migrants Home
- Budget 2020
- Jharkhand Votes
- The Federal Investigates
- The Federal Impact
- Vanishing Sand
- Gandhi @ 150
- Andhra Today
- Field report
- Operation Gulmarg
- Pandemic @1 Mn in India
- The Federal Year-End
- The Zero Year
- Premium
- Science
- Brand studio
- Home
- NewsNews
- Analysis
- StatesStates
- PerspectivePerspective
- VideosVideos
- Entertainment
- ElectionsElections
- Sports
- Loading...
Sports - Features
- Budget 2024-25
- BusinessBusiness
- Premium
- Loading...
Premium

Family portrait of novel coronavirus reveals a new type in India
While largely the coronavirus has been classified into two superclades, A and B, referring to Europe and South Asian origins respectively, a new variant has been found in India

Like every living organism, the novel coronavirus is undergoing mutation, a subtle change in the genomic sequence. Roughly, one generation of novel coronavirus lasts for about 10 hours. Obviously, the pace of the genetic mutation is much faster than what we observe in humans. It is said novel coronavirus mutates once in fifteen days. Each mutation is a new variant. Hundreds of variants have...
Like every living organism, the novel coronavirus is undergoing mutation, a subtle change in the genomic sequence. Roughly, one generation of novel coronavirus lasts for about 10 hours. Obviously, the pace of the genetic mutation is much faster than what we observe in humans.
It is said novel coronavirus mutates once in fifteen days. Each mutation is a new variant. Hundreds of variants have been identified so far. Phylogenetic analysis of genome from India reveals a hitherto unknown new type, accounting for whopping 41.2% of the sampled patients.
To put unfounded speculations at bay, a cautious CSIR tweeted: “There is no data for us yet to say that the new virus population (Clade A3i) among Indians is any more or less dangerous than the other virus population (Clade A2) present in India.”
Phylogenetic tree
A family tree can be constructed with grand grandparents and their offsprings, grandparents and their children, parents and their kin, we and our siblings. The tree of genealogy shows how the descendants are related to ancestors.
Biologists use a technique called a phylogenetic tree, to represent the evolutionary relationships among organisms and their mutant variants. The pattern of branching in a phylogenetic tree reflects how a particular variant evolved from a series of common ancestors. A phylogenetic tree of the variants of the novel coronavirus can be constructed to show the evolutionary connections between the descendant mutant virus and its parent.
Like we classify lion, tiger and cat having the same evolutionary pathway into one big happy cat family, the novel coronavirus is categorised into two big superclades ‘A’ and ‘B’. The former is called ‘European clade’, as mostly these mutations took place in Europe, and the latter ‘South Asian’ where these mutations took place before spreading worldwide. Further, these were classified into ten clades, A1a, A2, A2a, A3, A6, A7, B, B1, B2 and B4.
Branching out
Indian scientists from the Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, and Institute of Genomics and Integrative Biology (CSIR-IIGB) at Delhi took genomes of virus sampled from India and were preparing the phylogenetic tree. On carefully looking at the genomic sequence, they were assigning each of the 361 sequenced genomes in one or other clades — a simple enough task.
The initial two samples from Kerala, taken way back in early March, fell as expected into the B clade. These were individuals who had returned from Wuhan. The B clade did not spread much further, perhaps due to excellent containment. The B4 clade, a sub-type of the superclade B with potential origins from either East Asia or Oceania, was also found but limited to Gujarat and West Bengal.
The vast majority of the genome falls under the A superclade. This too was anticipated. After all, most introduction to India took place from Europe and the USA. The globally predominant clade A2a accounted for 45% of all the genomes. Curiously many samples collected from Ladakh belonged to A3 clade, mostly reported from Iran. Few belong to A1a clade, and the samples are mainly from Delhi. Nothing shocking or none startling.
Unexpected discovery
But a small nagging qualm remained. The five clades, A1a, A2a, A3, B and B4, found in Indian samples accounted only for about 57% of the total genome. What about the rest? They have hitherto relegated an uncategorised grouping, ‘others’.
Yet in the phylogenetic tree of virus variants sequenced from India, many genomes until now brushed aside as ‘others’, clearly could be seen to be falling into a new cluster. The genomic analysis also showed that the A3i clade stood out from other clades due to differences at four different places in its sequence.
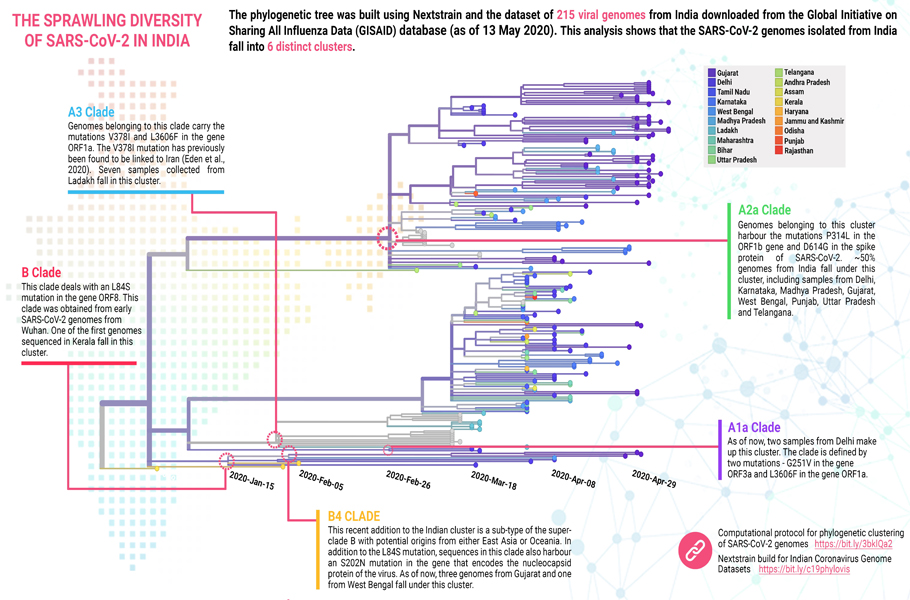
Researchers named it clade I/A3i. This new cluster alone accounted for 41.2% of all genomes sequenced in India. The second-largest variant of a novel coronavirus in India.
Of the samples from Tamil Nadu, Telangana, Maharashtra and Delhi, this clade is more than the A2a clade. In Bihar, Karnataka, Uttar Pradesh, West Bengal, Gujarat, and Madhya Pradesh newly characterised I/A3i is significant. However, until now in states of Haryana, Jammu and Kashmir, Madhya Pradesh, Odisha, and Rajasthan, this type of virus is least.
Globally 3.5% of genomes, which till date could not be mapped to any distinct known cluster, fall in this newly defined clade. Other than India, this clade is found in good numbers in Singapore and trace levels in a few other countries.
What we know about Clade I/A3i
Using the average mutation rate of the virus as a molecular clock, the phylogenetic study of all the genomic sequences collected from India shows that the most recent common ancestor of all the variants is around December 11, 2019. This agrees with all earlier studies and also puts to rest rumours that the Chinese suppressed more initial outbreak.
The same technique was also used to compute the origin of I/A3i clade as well as the A2a clade. The analysis of the predominant A2a show that it emerged around January 2, and the I/A3i on February 8. A traveller returning to Telangana from Indonesia, sampled on March 16, 2020, is the earliest person to have this type of viral genome.
A young tree will have fewer branches while the old tree many. If we start the family tree from great great grandparents and another from great grandparents, the number of branches in the second tree will be far lesser as compared to the first. The phylogenetic tree of I/A3i is short, suggesting that the origin and spread of the cluster were possibly from a single outbreak.
Of those infected by clade I/A3i virus in the sample, 69.4% were male, and 30.6% were female. In the case of the A2a cluster, 54.2% male and 45.8% female. The mean age of those infected by I/A3i virus is 36.8 years while for A2a, it is 41.1 years. Statistically, clade I/A3i appears to affect even younger people as compared to other clades. Nevertheless, the researchers did not find a significant difference with regards to the sex of the patient.

Does infection by I/A3i virus make one more sick or less? Are there any identifiable clinical features of those infected with I/A3i and other clades? Researchers say that clinical characteristics were not found to be significantly different between Clade I/A3i and the other clades.
The scientists have also identified four variants within this clade. Of these four, one mutant G11083T is silent and hence of no consequence. However, the other three, C6312A, C13730T and C28311T mutants have amino acid substitution. This means one of the proteins produced by these three mutant variant virus would be different from the ancestral one.
The mutations in I/A3i clade impact the genes related to the structural proteins of the virus, Nucleocapsid (N) and Envelope (E) genes. In A2a, the globally predominant clade, the mutations impact the genes connected with the Spike (S) and Membrane (M) genes using which the virus gains entry into the human cell.
When all genome sequenced in India is considered as a whole, the mutation rate is 1.64 × 10^−3 substitutions per site per year. However, when only the genomes from the A2a clade alone were taken, the estimated mutation rate was 1.65 × 10^−3. For A3i it was 1.4 × 10^−3 variants per site per year. In a nutshell, the A3i clade mutates slowly compared to the A2a clade. A slow mutation is good news for us but is disadvantageous for the virus. However, the researchers caution that more data and time is needed to see if this is actually the case.
The pre-print version of this study, A distinct phylogenetic cluster of Indian SARS-CoV-2 isolates, is authored by Sofia Banu, Bani Jolly, Payel Mukherjee, Priya Singh, Shagufta Khan, Lamuk Zaveri, Sakshi Shambhavi, Namami Gaur, Rakesh K Mishra, Vinod Scaria, Divya Tej Sowpati from CSIR-CCMB and CSIR-IGIB is uploaded in biorxiv and yet to be peer-review certified.
(The author is a science communicator with Vigyan Prasar)
