
- Home
- India
- World
- Premium
- THE FEDERAL SPECIAL
- Analysis
- States
- Perspective
- Videos
- Sports
- Education
- Entertainment
- Elections
- Features
- Health
- Business
- Series
- In memoriam: Sheikh Mujibur Rahman
- Bishnoi's Men
- NEET TANGLE
- Economy Series
- Earth Day
- Kashmir’s Frozen Turbulence
- India@75
- The legend of Ramjanmabhoomi
- Liberalisation@30
- How to tame a dragon
- Celebrating biodiversity
- Farm Matters
- 50 days of solitude
- Bringing Migrants Home
- Budget 2020
- Jharkhand Votes
- The Federal Investigates
- The Federal Impact
- Vanishing Sand
- Gandhi @ 150
- Andhra Today
- Field report
- Operation Gulmarg
- Pandemic @1 Mn in India
- The Federal Year-End
- The Zero Year
- Science
- Brand studio
- Newsletter
- Elections 2024
- Events
- Home
- IndiaIndia
- World
- Analysis
- StatesStates
- PerspectivePerspective
- VideosVideos
- Sports
- Education
- Entertainment
- ElectionsElections
- Features
- Health
- BusinessBusiness
- Premium
- Loading...
Premium - Events

Covid-19: Why it's so difficult to make antiviral drugs
Destroying viruses is elementary. Keeping host cells alive while we do it is the hard part. That is the critical challenge of developing an antiviral drug.

Remdesivir, made by Gilead Sciences, is the only drug approved by USFDA for the treatment of Covid-19. The molecule disrupts the replication of the new coronaviruses and slows the infection. Endorsed by bedside medical caregivers worldwide, it is one of the widely used drugs for treatment. The drug regimen requires multiple intravenous infusions, and hence, is given only to hospitalised...
Remdesivir, made by Gilead Sciences, is the only drug approved by USFDA for the treatment of Covid-19. The molecule disrupts the replication of the new coronaviruses and slows the infection.
Endorsed by bedside medical caregivers worldwide, it is one of the widely used drugs for treatment. The drug regimen requires multiple intravenous infusions, and hence, is given only to hospitalised patients. However, by the time the person is hospitalised, the virus has spread across the body. Remdesivir is often too late to do much good.
However, unseating it from the pedestal, the World Health Organization (WHO) in November 2020 announced, based on results of four trials, that Remdesivir “has not helped in curtailing mortality or reducing the need for mechanical ventilation among hospitalised COVID-19 patients”. At best, the available evidence suggests that Remdesivir does not seem to save lives but speeds recovery in those who do get well.
Surely, we need more antivirals to be able to choose from. So why don’t we have them? While 10-odd vaccines having been developed, why is it that the antivirals for treatment are so far behind.
The short answer is that finding safe antivirals is not that easy.
The ways of the virus
Studies have shown that it takes about 10 minutes on average for the novel coronavirus to fuse with a cell and infect it. However, within 10 hours of infecting a healthy cell in the respiratory tract, thousands of copies of the new virus burst out of it. While some of them will go on to infect the nearby lung cells, others mix with the air we breathe and come out to seek new hosts.
If the immune response is not swift and robust, the virus spreads far and wide quickly. One estimate says that a typical COVID-19 patient at peak infection would be teeming with one billion to a hundred billion virus copies in their body.
At that time, about one in a lakh cells in our respiratory system would be infected with the virus. An antiviral drug must reach all those cells and put a spanner in the works of the virus. Fashioning an antiviral requires an intricate understanding of the life cycle of the virus.
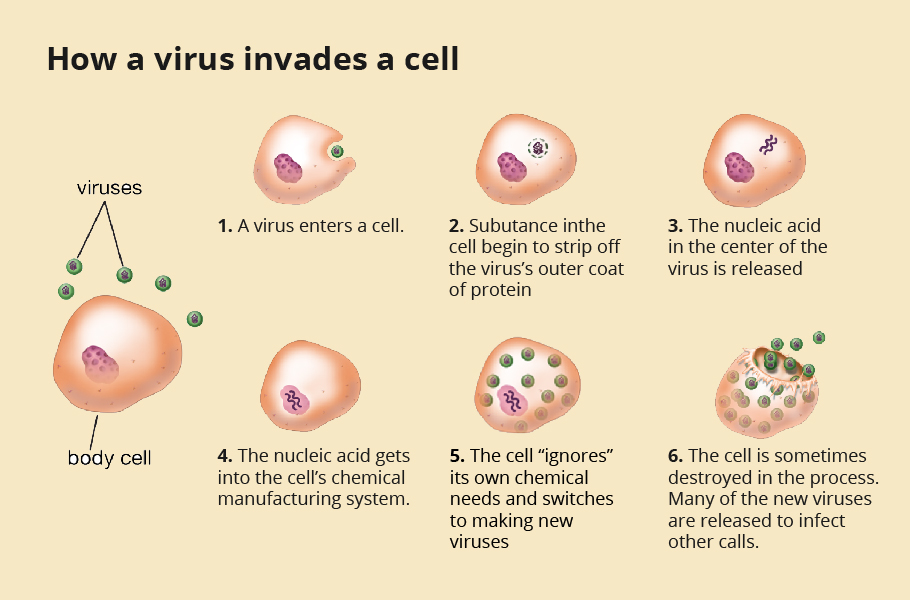
The life cycle of the virus
The journey of the virus begins with it taking a foothold onto a host cell. As we inhale the novel coronavirus, it enters into the respiratory tract. Like a lock and key, a specific structure, receptor-binding domain (RBD), on the spike protein of the novel coronavirus has evolved to bind effectively with the Angiotensin-converting enzyme 2 (ACE2) receptor found on many human cell surfaces.
Lungs, heart, blood vessels, kidneys, liver and gastrointestinal tract are rich in ACE2 receptors, making these cells prime targets for the novel coronavirus. The spike protein of the virus hooks onto the ACE2 receptor present in the epithelium in the nose, mouth, and lungs, particularly the alveoli. ACE2 is higher in patients with hypertension, diabetes and coronary heart disease, making them more susceptible to the SARS-COV-2 infection.
Manufacturing the viral proteins
Once the virus locks onto the ACE2 receptor, it fuses with the cell surface and slowly enters it. Once inside the cell, the virus sheds its coat and releases its genome, Ribonucleic acid (RNA). The genome, like a blueprint, is coded with a set of instructions for making copies of the virus. The first two-thirds of its 30,0000-alphabet-long genome codes for instructions to replicate its RNA. The remaining one-third of the genome code for manufacturing copies of its essential four structural proteins.
The tiny molecular machinery called ribosomes in our cells can read these genetic codes on the RNA. Like flowers of different colours stringed together to make a colourful garland, a chain of amino acids goes into making a protein.
The unsuspecting ribosome reads the genomic recipe and fastens various amino acids into a chain producing multiple viral proteins. Several ribosomes are recruited and simultaneously manufacture numerous copies of the structural proteins and copies of the genome critical to forming new viruses.
Copying the RNA
The human ribosome can read and makes thousands of copies of the proteins encoded in RNA. But this cellular machinery is not geared to make a copy of the RNA. Therefore, the RNA viruses, including SARS-COV-2, carry an instruction in their genome to produce a unique protein called polymerases.
The ribosome synthesizes the polymerase viral protein. Using the viral RNA as template, the polymerase builds a copy of the RNA by adding individual building blocks of the genome, nucleotides, one by one.
Once structural proteins and RNA genome are manufactured, they are assembled into a new virus at the pancake-like Golgi complex. A typical novel coronavirus needs about 1000 copies of nucleoprotein, 20 copies of envelope protein, hundred copies of spike protein, 2000 copies of membrane protein along with one copy of its RNA to become a complete virus particle.
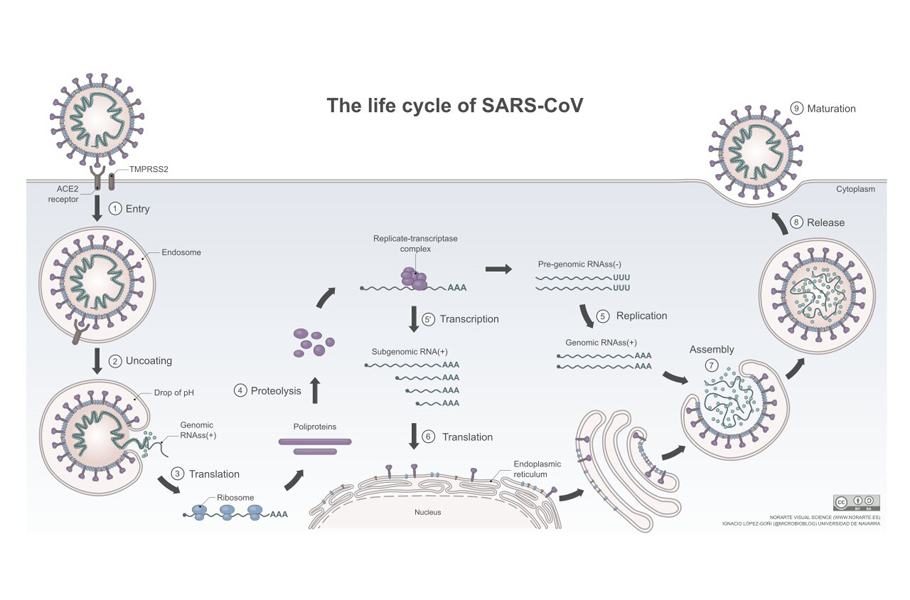
In this complex, the manufactured viral parts are sorted, packaged, and are wrapped by a tiny piece of the membrane to provide the new virus with a lipid envelope. The newly-formed viruses are then stuffed into vesicles, which make their way to the cell surface.
The fully assembled virus then exits the cell and seeks other healthy cells and repeats the replication process again.
Race to develop antivirals
The quest for a potent antiviral against the novel coronavirus commenced immediately after its destructive nature was understood. Discovering a new drug is rather a tedious, time-consuming process, mostly a hit and trial method. Once the pandemic raged, there was no time.
Scientists all over the world pored over available drugs to find antiviral activity against SARS-CoV-2. This is commonly done and is called repurposing a drug. More than 90 drugs, including ivermectin, hydroxychloroquine, chloroquine, doxycycline, azithromycin, favipiravir and lopinavir, were identified and examined for their clinical potential.
An unprecedented global human clinical trial was initiated, Solidarity trials, under the aegis of WHO, were conducted Initially, some of these drugs showed tantalising promises with an effective cure. Each of these drugs targeted a particular aspect of the virus life cycle.
Blocking entry
How about checking the virus from fusing and stopping it at the doorstep? A carbonyl compound Camostat was seen as a potential drug to stop the viral fusion. This drug, marketed since 1985, was used for the treatment of some forms of cancer and other diseases. It also showed promising results against some viral infections.
Human lung cells were grown in a laboratory petri dish and treated with the Camostat. The study showed that the drug could effectively inhibit the virus fusion when these cells were infected with the SARS-CoV-2 virus. With buoyant hope, clinical studies were initiated at Aarhus University Hospital in Denmark. About 205 COVID 19 patients were treated with Camostat. Still, they showed no noticeable effects on the duration of hospitalisation or severity of the cases in the clinical settings.
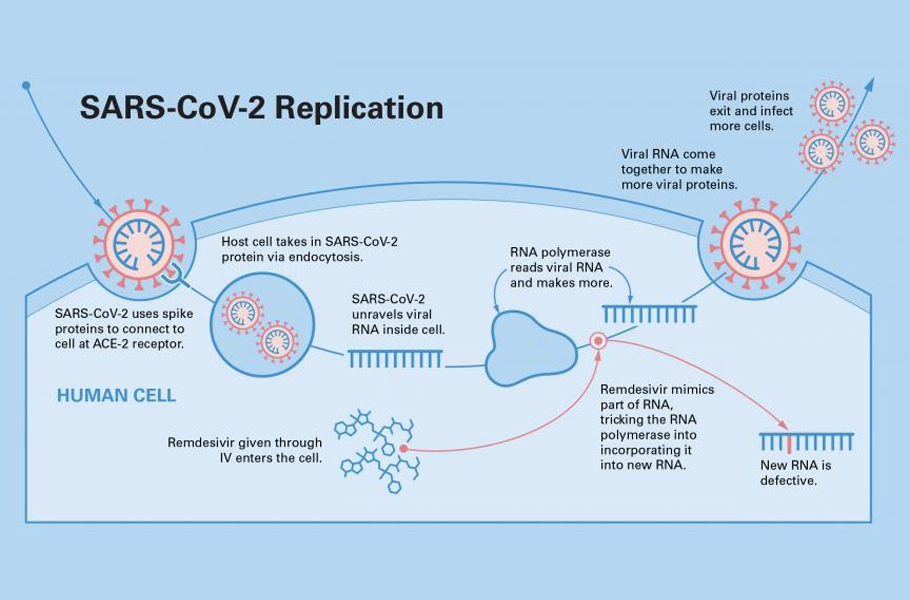
Desist undressing
If the virus cannot be halted at the entry-level, how about trying to prevent it from shedding its coat and releasing the RNA genome?
Enveloped viruses like SARS-COV-2 have to remove their outer coat to release their RNA into the cell. If a drug can hinder the un-coating, then it can be a helpful antiviral. Such an antiviral is potential prophylaxis, taken early to prevent the infection.
Influenza A virus, for example, is an enveloped virus. A viral protein called Matrix-2 protein is vital in letting the virus undress and release its RNA into the cell. Drugs like amantadine, rimantadine, zanamivir (Relenza) and oseltamivir (Tamiflu) jam the matrix-2 protein.
You cannot take off the coat without unfastening your buttons. With the matrix-2 protein jammed, the RNA cannot exit the envelop, viral replications do not occur, infection stalls. By arresting the virus from uncoating, the drugs slow the spread of infection and reduce the damage the infection causes.
Earlier research has shown that the Chloroquine-mediated inhibition of the hepatitis A virus was associated with resisting uncoating and preventing viral genetic shedding. The much-touted hydroxychloroquine was expected to stop the virus from removing its cloak, so that even after entering the cell, as the virus RNA remains encased, the entire replication cycle is blocked.
In studies conducted on animals such as monkeys and mice, hydroxychloroquine was encouraging. However, the randomised clinical trials unambiguously revealed that hydroxychloroquine did not help people with novel coronavirus infection nor prevented uninfected people from contracting the coronavirus.
Similarly, test tube studies with azithromycin were shown to block the coronaviruses replication in human cells. A clinical trial was conducted in the UK between April 7 and November 27, 2020, wherein 2582 patients were administered Azithromycin and 5182 patients were left to receive usual care. The study found no difference in duration of hospitalisation, invasive mechanical ventilation or death between the groups, implying no benefit of azithromycin in patients hospitalised with Covid-19.
Halting the replication
Can we disrupt the viral replication and restrain the spread of infection? Interfering with this vital copying mechanism of viral genes to form new viral genomes provides another promising approach to develop antivirals.
The interlocking pieces of a jigsaw puzzle can be assembled only when all the parts are correct. When a misshapen bit is used, the further assemblage of the puzzle is restrained. Likewise, suppose we can trick the viral polymerase into an inappropriate molecule rather than the right nucleoside, the resulting RNA chain will become gibberish junk.
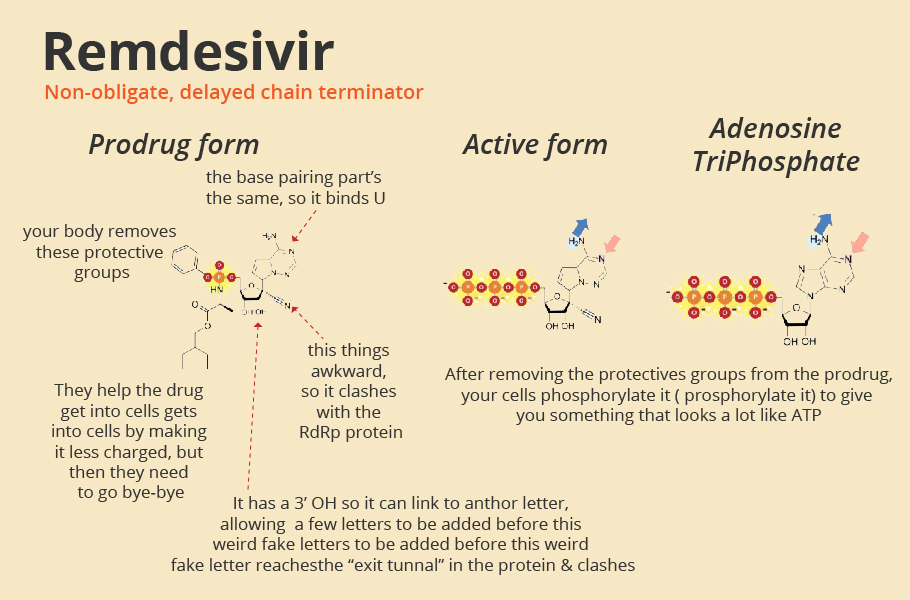
Drugs like Favipiravir, Remdesivir are decoy chemicals that resemble the building blocks of the RNA strand. The active form of favipiravir is mistaken by the polymerase protein to be a pseudo-purine. The drug molecules mimic both guanosine and adenosine during RNA replication.
The active form of Remdesivir resembles the RNA base adenosine. When a decoy molecule takes the place of nucleoside in the emerging RNA chain, no additional nucleoside can be joined to the chain due to a mismatch in the molecular structure. Thus, RNA replication halts midway.
The defective chain has to be discarded, and the viral polymerase has to start once again. The entire replication process slows down, and fewer mature viruses are assembled, slowing down the infection.
Often, viruses, in general, are poor at catching mistakes during replication. But not SARS-CoV-2. It has a proofreading mechanism encoded in its gene nsp14. This mechanism removes mismatched nucleotides during genome replication and transcription, making the drugs such as Favipiravir ineffective.
However, Remdesivir does not halt immediately after a decoy molecule is added. Perhaps it can partially sneak behind the proofreading mechanism of the virus. Nevertheless, even Remdesivir is not a match for SARS-COV-2, it seems.
Starving the virus
If nothing else works, why not deny vital energy and famish the virus? All activity requires energy. The energy for the cellular processes is obtained from D- Glucose. Glucose breaks down into few carbon compounds inside the cell, one of which is pyruvate anion (CH3COCOO-) with the release of energy. This metabolic process is called glycolysis.
2-Deoxy-D-Glucose, 2-DG in short, is a mimic of D-Glucose. The overworking viral infected cells are starved of energy and are on the lookout for Glucose. As the 2-DG impersonates D- Glucose, it can enter the infected cells. However, 2-DG is unfit to perform glycolysis, and no energy is produced. However, as the cell already has an adequate concentration of 2-DG, normal D-Glucose does not enter.
The infected cell is starved of energy; the cell and the viruses inside it die. The exact mechanism resulted in 2-DG being tested against cancers and tumours for about 20 years but is yet not an approved drug for anticancer treatment. Approved for emergency use now in India for anti-COVID-19 treatment, the drug has still not gone through standard randomised placebo controlled phase-3 clinical trials.
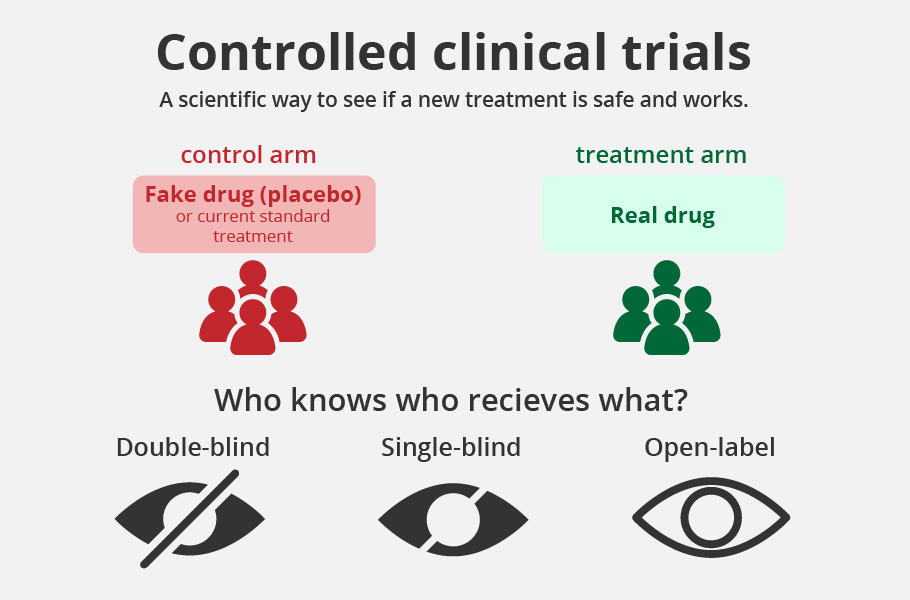
Is this drug a game-changer? We hope it is, but not likely. The limited Phase-3 unblinded clinical trial conducted with 220 patients showed that 42% who were given 2-DG and Standard of Care (SoC) improved symptomatically compared to 31% with SoC by Day-3.
The trial was not random or blinded with placebo control, the gold standard for drug trials. Further, the trial excluded critically ill patients suffering from asthma, interstitial lung disease, diabetes mellitus or any condition predisposing to hypoglycaemia.
That is, almost all those who are classified as comorbid conditions were not part of the trial. The drug should not be taken outside hospitalisation. How much this drug would help in tiding over the pandemic is yet to be seen.
Why antivirals are a challenge
Almost all the antivirals studied under the WHO Solidarity trial do not seem to be hopeful against novel coronavirus. These drug regimens had little or no effect on hospitalised patients with Covid-19, as indicated by overall mortality, initiation of ventilation, and duration of hospital stay, the study has concluded.
When we can make so many vaccines against the virus, why is there a dearth of antivirals?
Belonging to two different kingdoms of life, bacterial and human cells are very distinct features that make antibiotics possible. For example, bacterial cell walls are made of a polymer called peptidoglycan, and human cells do not have them. Penicillin and its derivatives like Amoxicillin, Ampicillin antibiotics inhibit the repair of the peptidoglycan layer.
When this drug is given, the bacterial cell wall disintegrates, and the pathogen dies. Hence, the antibiotics that prevent the synthesis of peptidoglycan can kill the bacteria without causing harm to the humans taking the drug.
In contrast, antiviral drugs have to target some essential aspect of the viral life cycle. At every step, the virus uses the host cellular machinery. Once a virus enters the cell, you can hardly kill the virus without destroying the infected cell.
Viruses as such offer no such easy targets. Targeting the replication process of the virus can be toxic to the human host as well.
Researchers hope that by knowing the intricacies of the SARS-CoV-2 virus and its interactions with human cells, we can identify unique features of the novel coronavirus in how it survives, replicates, and evades our immune surveillance. By exploiting its weakness, we hope to develop antivirals that put a spanner in the works without harming normal human cells.