
- Home
- India
- World
- Premium
- THE FEDERAL SPECIAL
- Analysis
- States
- Perspective
- Videos
- Sports
- Education
- Entertainment
- Elections
- Features
- Health
- Business
- Series
- In memoriam: Sheikh Mujibur Rahman
- Bishnoi's Men
- NEET TANGLE
- Economy Series
- Earth Day
- Kashmir’s Frozen Turbulence
- India@75
- The legend of Ramjanmabhoomi
- Liberalisation@30
- How to tame a dragon
- Celebrating biodiversity
- Farm Matters
- 50 days of solitude
- Bringing Migrants Home
- Budget 2020
- Jharkhand Votes
- The Federal Investigates
- The Federal Impact
- Vanishing Sand
- Gandhi @ 150
- Andhra Today
- Field report
- Operation Gulmarg
- Pandemic @1 Mn in India
- The Federal Year-End
- The Zero Year
- Science
- Brand studio
- Newsletter
- Elections 2024
- Events
And the Chemistry Nobel goes to… Deciphering protein structures and designer proteins

David Baker of the University of Washington, Seattle, WA, USA and Howard Hughes Medical Institute, USA, received half of this year's Chemistry Nobel prize for "computational protein design”. At the same time, Demis Hassabis and John M. Jumper of Google DeepMind, London, UK, shared the other half for "protein structure prediction”.Proteins - a basic primerThe term 'protein' conjures images...
David Baker of the University of Washington, Seattle, WA, USA and Howard Hughes Medical Institute, USA, received half of this year's Chemistry Nobel prize for "computational protein design”. At the same time, Demis Hassabis and John M. Jumper of Google DeepMind, London, UK, shared the other half for "protein structure prediction”.
Proteins - a basic primer
The term 'protein' conjures images of pulses, eggs, and meat. Protein is frequently described as a nutrient in school science textbooks alongside carbs, lipids, vitamins, and minerals.
However, proteins are much more than just a nutritional category. They are the workhorses in cells, performing most of the work and being critical for constructing, functioning, and controlling the body's tissues and organs.
Enzymes (such as pepsin, which helps break down proteins in meat, eggs, seeds, and dairy products into amino acids) help catalyse chemical reactions, hormones (such as Insulin, which enables the body metabolise and use energy from nutrients), proteins (such as XPA protein) help synthesising and repairing DNA, proteins (such as haemoglobin) assist in transporting materials across the cell, and proteins (such as G proteins) enable receiving and sending chemical signals.
Proteins are made up of small molecules known as amino acids. A human cells has 80,000 to 400,000 proteins, comprising only twenty different types of amino acids. Just as a garland is made by arranging several flowers in a specific order, the chain of amino acids in a different arrangements produce diverse proteins. Proteins are often called macromolecules to distinguish them from smaller size molecules, such as glucose, water and so on.
Some proteins are made of a low number of aminoacids; for example, Insulin contains just 51 amino acids in its chain. An average adult's pancreas contains roughly 200 units of Insulin, which is about 70 grams. Titin1, also known as connectin, is the biggest known protein. It contains between 27,000 and 35,000 amino acids. The adult human body has around 500 gram of titin. Titin accounts for approximately 10% of muscle mass and is the third most abundant protein in muscle, behind actin and myosin.
Structure of proteins
You usually get one piece when you order dosa, but when you order idli, you get two. An idli vada combination typically includes two ildis and one vada. As is the situation with proteins.

Proteins are macromolecules made from string of amino acids arranged in a particular order.
Let us look at myoglobin, a tiny, brilliant red protein, that gets its colour from the iron atom it binds, found in muscle cells that give meat its red colour. It stores oxygen using the iron atom in the muscles and releases it when we perform strenuous physical effort. The protein is a single chain of 153 amino acids, similar to a pearl necklace. Proteins made from single chain of amino acids is called a monomer as it functions by itself. Monomers are like dosa, they come single chain.
However, haemoglobin is a globular protein composed of four chains of amino acids which come together forming a complex. Unless they come together correctly they do cannot function. In adults, of the four chains two of them are called alpha chains, while the other two are called beta chains. In newborns foetal hemoglobin has the same two alpha chains but other two are different called gamma chains. But in adults and newborns the hemoglobin molecules have the same structure. Proteins like hemoglobin are like idli-vada combo. There are more than one monomer in the final product.
Protein folds
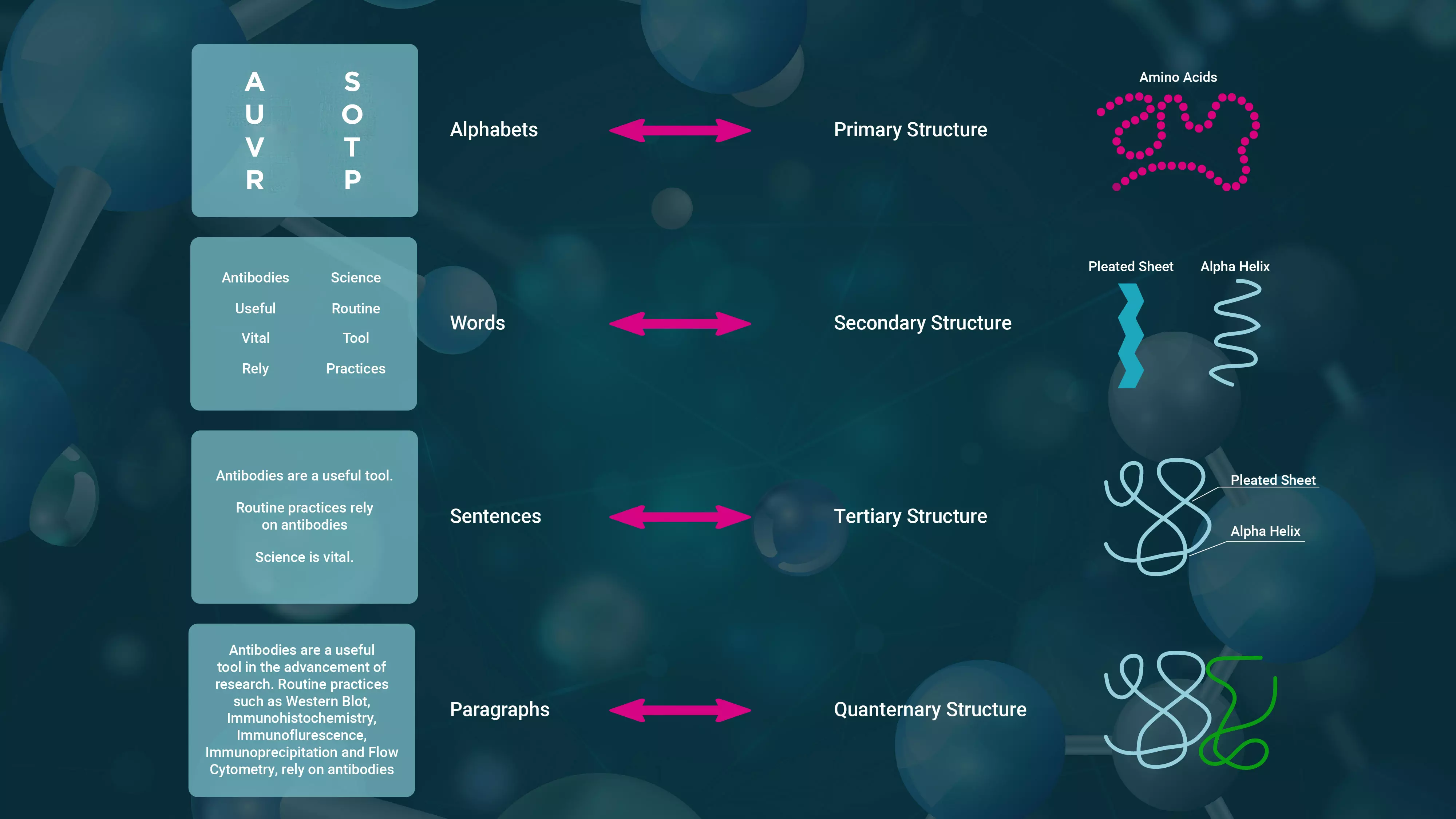
A key is just a piece of metal. The pattern of notches and teeth determines if it is a key for a corresponding lock. The key shaft must fit correctly into the lock cylinder, and its thickness and length must correspond to the lock. Likewise, the correct 3D structure of proteins is critical to their biological activities.
If you leave a piece of paper for a few days, it will naturally curl. Similarly, an amino acid chain's sequence folds into a repeating pattern. Alpha helices and beta sheets are the most prevalent stable folding patterns in the set of secondary structures in proteins. Alpha helices resemble coiled crepe paper streamers, while beta sheets resemble saree pleats. One section of the amino acid sequence folds spontaneously into an alpha helix, while another forms a pleated sheet. Thus, a chain may contain many helices, sheets, and other patterns.
When folding a shirt, we fold it in half vertically and then horizontally and make sure to tuck the sleeves inside the fold to make it compact. When we fold a pant, we fold it half vertically at the crotch, then half horizontally at the legs, and then fold it farther to make it compact. Similarly, the secondary structure of proteins folds into a compact three-dimensional form known as the tertiary protein structure. Sometime tertiary structure can function as a monomer. For example, myoglobin, a monomer, which has 154 amino acids, first folds into eight alpha helices linked by loops. The whole protein folds into a tight globular form. Because of this particular fold, a placeholder for the iron that binds the oxygen is formed in the core, allowing it to store and release oxygen.
A shirt or pant is folded and kept separately, but a salwar kameez set or sari with matching blouse is folded and tucked inside to store as a set. Similarly, specific proteins may have many chains, known as subunits. The haemoglobin, for example, has four subunits: two alpha and two beta chains. Insulin comprises two subunits: A, which has 21 amino acids and two tiny alpha helices, and B, which contains 30 amino acids and one alpha helix. Thus in insulin two monomers have to combine, only then it will function. Eventually, the entire structure transforms into a tiny spherical shape like an oil drop in water.
In certain proteins monomers need to form complexes with other monomers or other molecules to function. These are called quaternary structures. Insulin has a quaternary structure, like a idli-vada combo, whereas myoglobin is a single monomer, like a piece of dosa.
Structure and function
Let us start with simple water: one oxygen and two hydrogen atoms - H2O. The solitary electron from both hydrogen atoms is lent to oxygen to create the molecule. As a result, without an election, hydrogen is slightly positive, whereas, with extra electrons, oxygen is somewhat negative. As a result, the hydrogen atoms repel each other, causing them to be 104.5° apart and giving the structure a peculiar micky mouse form. Now, slightly negative oxygen may attract hydrogen from another water molecule. In contrast, positive hydrogen can attract oxygen from two separate water molecules. Thus, water molecules in a container create a weak bond with surrounding molecules, giving water its characteristic features.

Due to mutation, if one or more amino acid change, the structure of the protein is greatly affected; the misfolded protein at times may cause serious diseases.
The protein's three-dimensional tertiary structure also gives particular features. For example, specific folds form a placeholder for the iron that binds the oxygen in the middle of globular myoglobin, allowing it to store and release oxygen. In contrast, collagen is a lengthy protein comprising many amino acid chains twisted together like a rope or cable. The rope-like stiff characteristic of collagen provides strength to the tendons and ligaments that link bones and muscles. Porin protein, on the other hand, is mostly beta pleats and has the form of a cylindrical tube with open ends, allowing it to function as a channel for a tiny chemical to diffuse across a cell membrane.
Knowing the shape
Knowing whether the end of the pencil is sharp or blunt allows us to predict its behaviour. Similarly, the structure and shape of the protein fold reveal information about how it will function. Histidine is the 98th amino acid found in healthy myoglobin. However, if it is replaced with Tyrosine through mutation. Due to this mutation, what amoni acids appear in the outer layer changes, which makes them stick together and forms lumps, like rice cooked with too much water gets sticky and aggregated. They cannot store oxygen as effectively, resulting in myoglobinopathy muscle disease. Similarly, changing the sixth amino acid in the beta chain of the beta-hemoglobin gene from normal glutamic acid to valine causes significant alterations in the protein's behaviour. Healthy red blood cells are typically laddu-shaped, but aberrant ones formed by mutant proteins are sickle-shaped.
Scientists utilise the X-ray crystallography technique to determine the 3-dimensional structure of the protein. They direct X-rays at the crystallised protein. X-rays bounce off, or scatter, from the atoms within the protein. The scattering produces a pattern of bright and dark spots on a detector. Just as we may predict the form of an item by looking at its shadow, scientists use computers to study the unique pattern of variation of brightness of the spots generated on the detector to establish the position of each atom in the crystal. They then can generate a 3D representation of the protein's structure.
In the 1950s, Cambridge researchers John Kendrew and Max Perutz succeeded in decoding the three-dimensional structures of myoglobin and haemoglobin using X-ray crystallography, for which they received the Nobel Prize in Chemistry in 1962. Since then, researchers have meticulously determined the structure of approximately 200,000 proteins.
It has been recognised that the sequence of amino acids in a protein and the environment around it determines its three-dimensional structure. A specified protein sequence given the same environment will fold in one way and not in another. Given an amino acid chain, it should be easy to anticipate its form when folded.
However, determining the protein fold was a significant hurdle. Nowadays, various experimental approaches, including X-ray crystallography, Nuclear Magnetic Resonance (NMR) spectroscopy, and cryo-electron microscopy, are employed to identify the structure. They take anywhere from a day to a year and are expensive.
AI and protein structure
By the 1990s, artificial intelligence had succeeded in image identification and facial recognition. Researchers attempted to apply AI methods to identify patterns in protein folding and underlying amino acid sequences. Several studies provided predictions; however, how can you test the validity? In 1994, researchers launched a biennial competition named 'Critical Assessment of Protein Structure Prediction (CASP)' to evaluate protein predicting methods based on their accuracy. The organisers used a group of proteins whose amino acid sequences were known, however while those structures had been established using X-ray crystallography they had not yet been released publicly. They kept the structures a secret and challenged other researchers who were not involved in the structure determination to predict it using the sequences.

AlphaFold2 achieved great success in computing the structure of monomer proteins.
Demis Hassabis co-founded DeepMind, a firm that created AI applications for popular board games, including chess, shogi, and Go, in 2010. He enrolled for the thirteenth CASP competition in 2018. Hassabis' team was able to anticipate the structure with over 60% accuracy using a type of neural network based AI model. They called it AlphaFold. However, they were at a loss because they were unable to enhance their accuracy no matter what they did.
John Jumper, fascinated with protein dynamics, joined Google DeepMind's research team in 2017. Jumper and Hassabis co-led a new venture that used the newest neural networks known as transformers to detect patterns in sequences of amino acids of the 2,00,000-odd proteins whose structures have already been decoded. The much-upgraded version AlphaFold2, published in 2020, was a huge success for monmeric proteins. In the 2020 CASP competition, AlphaFold2's accuracy was virtually identical to X-ray crystallography structures.
Designer proteins
David Baker also competed in the CASP competition. He created another neural network based AI program named Rosetta to predict protein structure and entered the 1998 competition. Even though his algorithm performed relatively well, he decided to try something many had been not able to do successfully. Instead of trying to find the structure from the amino acid sequence; they attempted to find what sequences would yield a given structure like different clothes that fit a tailors dummy. He modified Rosetta to achieve this goal and acquired predictions for amino acid sequences for a desired protein structure.
When a structure was provided, Rosetta first recognised its constituent parts before searching a database of all known proteins for a small piece that matched the detected components. The program then determined the amino acid sequences for that component. As a result, the amino acid sequences for the appropriate protein structure was found in stages.

De novo protein Top7 has 93 amino acids which folds into two alpha helices packed on a five beta sheets.
Baker set out to see if a particular sequence that was found by pairing sequences to the designed structure resulted in the required protein structure. His team synthesised the DNA sequence for the required amino acid sequence. They put the DNA into bacteria using genetic engineering and were able to produce the resulting protein His team utilised the age-old technique of X-ray crystallography to determine if the structure of the protein they had produced is identical to the predicted one. They found the one they called Top7, has a nearly similar structure to the one they had designed initially. Thus, Baker demonstrated that functional designer proteins, or proteins with desired features, may be created. These designer proteins are known as 'de novo proteins' as they do not exist in nature and are created using the structure as a guide
Controversy
Baker has made Rosetta's code available as open source for researchers. Google DeepMind has made the AlphaFold2 code publicly available, and over two million people from 190 countries are using it to detect protein structures and conduct drug development research.
However, in May 2024, Google's DeepMind and Isomorphic Labs (a subsidiary of Google Group's parent firm, Alphabet) disclosed AlphaFold3, which works for complexes, as a closed-source technique, prompting condemnation from scientists worldwide. "The AlphaFold3 case is a cautionary tale. While private money might speed scientific advancement, it must not come at the price of open research," says S Krishnaswamy, a retired professor at Madurai Kamaraj University's School of Biotechnology and also in All-India People's Science Network.
DeepMind did not reveal the whole code or the model's inner workings; instead, it provided a simplified algorithm description and a web server for restricted use. "Companies have a valid interest in protecting their investments, but excessive IP restrictions can hinder scientific progress and limit the societal benefits of innovation", according to him. "The limits of AlphaFold3 in terms of access present ethical problems. Restricted access has the potential to limit AlphaFold3's use for drug development to well-funded universities and enterprises. This might drastically slow the development of life-saving therapies, especially for illnesses affecting impoverished countries," he noted.